I Registration Categories
1) New chemical entity never marketed in any country.
i. Drug substance and its preparations made by synthesis or semi-synthesis.
ii. New chemical monomer (including drug substance and preparation) extracted from natural sources or by fermentation.
iii. Optical isomer (including drug substance and preparation) obtained by chiral separation or synthesis.
iv. Drug with fewer components derived from marketed multi-component drug.
v. New compound drugs.
vi. A preparation already marketed in China but with a newly added indication not yet approved in any country.
2) Drug preparation with changed administration route and not marketed in any country
3) Drug marketed ex-China, including:
i. Drug substance and its preparations, and / or with changed dose form, but no change of administration route.
ii. Combination preparations, and / or with changed dose form, but no change of administration route.
iii. Preparations with changed administration route and marketed ex-China.
iv. A preparation already marketed in China but with a newly added indication approved ex-China.
4) Drug substance and its preparation with changed acid or alkaline radicals (or metallic elements), but without any pharmacological change, and the original drug entity already approved in China.
5) Drug preparation with changed dose form, but no change of administration route, and the original preparation already approved in China,
6) Drug substance or preparation following national standard.
II Application Dossier Items
A Summary
1) Name of the drugs.
2) Certified Documents.
3) Objectives and basis for R & D.
4) Summary of main study work.
5) Draft of packaging insert, note to the draft, and latest literature.
6) Design of packaging and labeling.
B Pharmaceutical data
7) Summary of Pharmaceutical Study,
8) Research information and relevant literature of the production process of the drug substance, research information and relevant literature of formula and process of the preparations.
9) Study information and relevant literature for the chemical structure and components determination.
10) Study information and literature for quality specification.
11) Draft of quality specification and notes, and providing reference standard.
12) Test report of drug sample.
13) The source, test report and quality specification of drug substance and adjuvants.
14) Stability study and relevant literature.
15) Selection basis and quality specification of immediate packing material and container.
C Pharmacology and toxicology study information.
16) Summary of pharmacology and toxicology study.
17) Primary pharmacodynamics study and literature.
18) General Pharmacology study and literature.
19) Acute toxicity test study and literature.
20) Long -term Toxicity study and literature.
21) Special safety study and literature of hypersensitive (topical, systemic and photo-toxicity), hemolytic and topical irritative (blood vessel, skin, mucous membrane, and muscle) reaction related to topical and systemic use of the drugs.
22) Study and relevant literature on Pharmacodynamics, toxicity and pharmacokinetics change caused by the interactions amongst multiple components in the combination products.
23) Study and literature of mutagenicity test.
24) Study and literature of reproductive toxicity.
25) Study and literature of carcinogenicity test.
26) Study and literature of drug dependence.
27) Study and literature of non-clinical pharmacokinetics.
D Clinical Study Information
28) Summary of global clinical study information.
29) Clinical study protocol.
30) Investigator’s Brochure.
31) Draft of Informed Consent Form, approval of the Ethics Committee.
32) Clinical study report.
III Notes to Application Information Items
& nbsp;1) Information Item 1, Name of the drugs, includes International Nonproprietary Name (INN), Chemical Name, English Name, and Chinese Phonetics. Chemical structure, Molecular Weight, Molecular Formula shall be noted. The Nomenclature of the drug should be explained for any new name.
2) Information Item 2, Certified Documents, includes,
i. Certified Documents of lawful registration of the Applicant (such as business license), copies of Drug Manufacturing License and update page, copies of GMP Certificate. For the application of production of new drugs, copies of GMP Certificate for the workshop where the sample product of the drugs was manufactured should be provided.
ii. Certified Documents stating patent status and ownership of this entity and formula, production process of the drug, and letter of guarantee stating that no infringement upon the patent rights of others.
iii. Copies of official approvals of the research proposal of narcotics, psychotropic and radioactive drugs.
iv. For the application of production of new drugs, copy of Approval of Clinical Study of New Drugs and the quality standard of investigational drugs should be provided.
v. For the application of production of preparation, certified documents to evidence the legal channels of drug substance should be provided, including copies of certified approval document of drug substance, drug standards, test report, business license of manufacturers of drug substance, Drug Manufacturing License, GMP Certificate, sales invoice, and supply contract.
vi. Copies of the Drug Packing Material and Container Certificate or Import Drug Packing Material and Container Certificate for the immediate packing material and container.
) Information Item 3, objectives and basis of the application, includes R&D, marketing status and related literature of the drugs, or the use and production status of the drugs, summary of study of the reansonaleness of the preparation and the necessity of clinical use, domestically and overseas.
4) Information Item 4, summary and evaluation of main research results, includes the summary of main research results by the Applicant, and a comprehensive analysis of safety, efficacy, and quality controllability of the drugs of the application.
5) Information Item 5, draft of insert sheet, notes to the draft and latest literature related, includes the sample of draft of packaging insert sheet drafted in accordance with the relevant regulations, notes on how each items of the insert sheet were drafted, and the latest relevant literature.
6) Information Item 7, Summary of Pharmaceutical Study, refers to the summary of experiment and global literature of Pharmaceutical Study of the drug in the application (synthesis process, selection of dosage form, screening of formula, determination of structure, quality study and determination of quality standards, and stability study). & nbsp;7) Information Item 8, research information of the production process of the drug substance, includes technology process and chemical reaction equation, initial raw material and organic menstruum, reaction conditions (temperature, pressure, duration, catalyst), operation procedure, refining method,main physical-chemical constants, data and results collected at various stages etc. The raw material input, and output yield, as well as possible impurities or other by-products produced or mixed during the production process should be explained. Research information of preparation formula and production process shall include information of starting materials, formula selection, production process and verification.
8) Information Item 10, experiments information and literature for quality research, includes physical-chemical properties, purity inspection, dissolution, content assay, methodology validation, data and results collected at various stages etc.
9) Information Item 11, draft of drug quality specification and notes, and reference standard shall be provided: Quality specification shall comply with the format of the current version of Chinese Pharmacopoeia, and the terminology and units of measure of Chinese Pharmacopoeia should be used. Reagents, reagent solution, buffer solution, titrant and others used and their concentration should follow the current version of Chinese Pharmacopoeia. In the event of a different one was used, detailed explanations should be provided. Reference standard shall be provided with separate information attached to explain the source, physical-chemical constants, purity, content, and measurement method and data of the drugs. Notes to the draft of drug standards shall include the selection of items to be controlled, selection of method, inspection and purity and limitation range, as well as the basis to decide each item.
10) Information Item 12, the test report of the sample products, means the self-test report of the sample products of the drugs in the application. Self-test report for at least one batch of sample product should be provided before the clinical study. Self-test reports of the consecutive 3 batches of sample products should be provided for the market authorization approval after completion of the clinical study.
11) Information item 14, experiments information and literature of the stability study of the drugs, includes study of the factors affected and stability test conducted together with the use of the immediate packing material and container.
12) Information item 16, Summary of pharmacology and toxicology study, refers to the summary of experiment and global literature of pharmacology and toxicology study of the drug in the application (including pharmacodynamics, mechanism of action, general pharmacology and toxicology, pharmacokinetics etc.).
13) Information item 27, summary and literature of non-clinical pharmacokinetics, refer to the summary of experiment and literature of non-clinical pharmacokinetics (animal) of the drug in application (absorption, metabolite, distribution, excretion)
14) Information item 28, Summary of global clinical study information, refers to summary of global literature, abstract and latest updating regarding the clinical trial of the drug in the application.
15) Information item 29, Clinical study protocol: Clinical study protocol should cover details to deal with the critical items including proposed indication, usage and dosage, which should be supported with submission of study information. Clinical study protocol should be scientific, complete and there should be a comprehensive summary of non-clinical and clinical information related to the key analysis of the potential risks and benefit of proposed trials.
16) Information item 29, Investigator’s Brochure, refers to summary of existing non-clinical and clinical information of the drug in the application, for the purpose to provide Investigator and other participators with information to aid them in understanding the characteristic of the drug and Clinical study protocol. Investigator’s Brochure should be concise and objective. Investigator’s Brochure should be conscious and impersonal.
IV Table of Application Information Item and Notes
A Table of Application Information Item
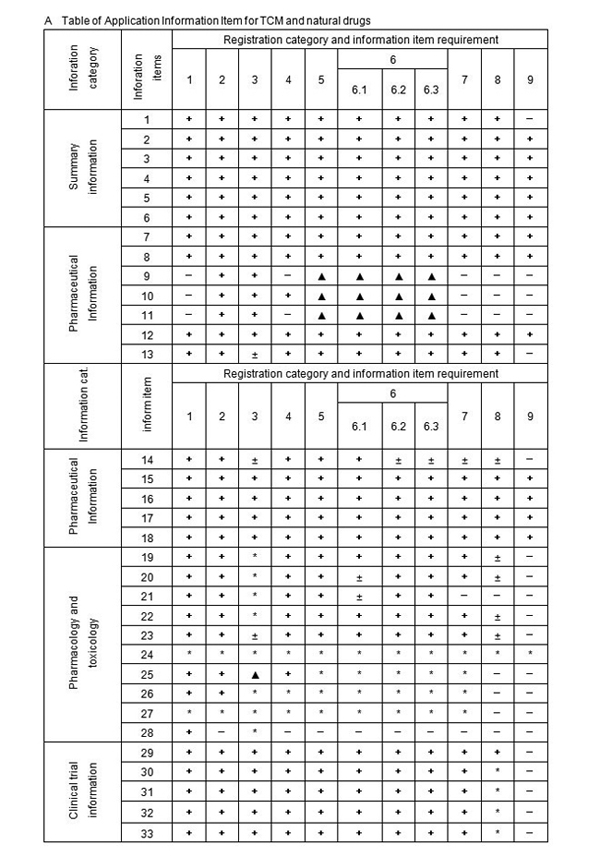
图片1
Notes: 1. + Denote the information must be submitted,
2. ± Denote literature can be used instead of test information,
3. − Denote the information may be exempted,
4. ∗ Denote the information shall be submitted according to the requirement, ∗6 refer to note 6.
5. △ denote that the provisions 4 of “V , Requirement For Clinical Study” shall apply.
6. literature refers to literature and / or summary of literature of all pharmacology and toxicology study information of the drug in the application (including pharmacodynamic, mechanism of action, general pharmacology, toxicology and pharmacokinetics)
B Notes
1) Information item 1-30 (except for item 6) of Table of Application Information Item should be submitted for Application of Drugs under Registration Categories 1-5. Re-compiled summary Information Items 1-6, Information Item 12 and 14, clinical Information Items 28-32 and other changes and supplemental information shall be submitted and numbered with the numbers of Information Items upon the completion of the clinical study. For the drugs under Registration Category 1, upon the completion of the clinical study, all the required information of Information Items 1-30 should be re-edited according to the result of trails conducted during clinical study, and then be re-submitted. When the registration of drug substance and registration of preparation of the chemical drug under Registration Category 3 and 4 are applied at the same time, the registration of drug substance should comply with the requirement for production.
2) Information Items 1-16 and 28-30 shall be submitted for the application for drugs under Registration Category 6 according to the requirement of the Table of Application Information Items. If the clinical study was required, upon completion of the clinical study, Information Items 28-32 and other changes and supplemental information shall be submitted and numbered with the numbers of Information Items.
3) During the registration of drugs under Registration Category 6, there should be a comprehensive quality study of the process and formula of the drug, and quality comparison with the already marketed drugs according to the national standards. When it is not possible to conduct the quality comparison with the already marketed drugs according to the national standards, a quality study should be conducted according to the requirement for registration of new drug, and if necessary, quality provisions in the national standard can be appended and /or revised.
4) During the application only for preparations, the lawful Certified Documents to evidence the lawful sourcing of the drug substances shall be provided in 2 duplicates, which should be respectively categorized into information item 2 (certified document) and information item 13 (The source, test report and quality specification of drug substance and excipient). For the applicant using domestic drug substance, the documents that should be provided include copies of certified approval document of drug substance, drug standards, test report, business licenses of manufacturers of drug substance, Drug Manufacturing Certificate, GMP Certificate, supply contract signed with manufacturers of drug substance and sales invoice. When the import drug substances were used, copies of supply contract signed with manufacturers of drug substance or its legal domestic agent, Import Drug Certificate, or Pharmaceutical Product Certificate, test report from Drug Control Institute of the local Customs, and drug standards shall be provided. During drug registration, use of investigative preparation with the drug substances without Import Drug Certificate, or Pharmaceutical Product Certificate must be approved by SFDA.
5) The reproductive toxicity research information corresponding to the drug used for the people at child-bearing age should be submitted based on the natures of the indications and characteristic of the new drug
6) For any of the drugs with expected treatment period longer than 6 months inclusive, or intermittently used for treatment of chronic and recurrent disease for a regular period of time, experiment information or literature on Carcinogenicity should be provided, and information of carcinogenicity test or literature should be submitted for the following new drugs, based on the indication and characteristic of action:
i. The chemical structure of the new drugs or the metabolite of the new drugs is similar to the known carcinogen.
ii. During long-term toxicity experiment, cytotoxic effects were shown or extraordinary activation on the growth of cells in certain visceral organs and tissues were caused. iii. Drug with a positive test result during mutagenicity test.
7) For new drugs acting on central nervous system, such as analgesics, depressants, stimulants, and drugs with chemical structure liable to cause human drug dependence, experiment information of drug dependence should be submitted.
8) For the new drug under Registration Category 1, toxicokinetics study should usually be conducted during the repeated doses toxicity study.
9) Under the Registration Category 1, the optical isomer obtained from a known drug through chiral separation or synthetic method and its preparation, the research information and relevant literature compared between racemate and mono-isomer in areas on pharmacodynamics, pharmacokinetics and toxicology (normally acute toxicity) should be provided to indicate the justification of the R&D. When the safety range of racemate is narrow, and the available information indicates that the unexpected toxicology (irrelevant to pharmacology) is considerably high, the toxicology test of mono isomer with repeated doses (normally lasting for 3 months) or other toxicology tests (such as reproductive toxicology) shall be provided based on the comprehensive information such as clinical course of treatment, dosage, and indications of the drugs, as well as the people using the drugs.
10) For drugs under Registration Category 1 with fewer components drugs derived from already marketed multi-component drugs, if there exists no substance explained in note 6 herein in the component, then Information Items 23-25 may be exempted.
11) For the new compound preparation under Registration Category 1, Information Item 22 should be submitted.
12) For the new compound preparation under Registration Category 1, information of toxicity test of repeated dosage compared with single dosage should be provided, and if no increase in toxicity indicated in the toxicity test of repeated dosage, and no change in the target tissue, Information Item 27 should be exempted.
13) For the new compound preparation under Registration Category 1, if there is no significant change in animal pharmacokinetic study results, then Item 23-25 should be exempted.
14) For the new drugs under Registration Category 2, the route of administration during the pharmacology and toxicology study should be the same with that to be used in clinical study. Generally, the pharmacokinetic test or the related toxicology study information (such as topical and repeated dose toxicity) compared with the original route of administration should be provided.
15) For the drugs under Registration Category 3, the preparations with change in route of administration and already marketed overseas, emphasis should be focused on the drug absorption or topical toxicity influenced by the excipients, and if necessary, the pharmacokinetic test or other toxicology study should be provided.
16) For the new drugs under Registration Category 4, pharmacokinetic, main pharmacodynamic, normal pharmacology and acute toxicity test information compared with the already marketed drugs should be provided to reflect the difference before and after the changes, and if necessary, the research information on repeated doses toxicity and other relevant pharmacology and toxicology study should be provided. If the preparation is made by changing the acidic or alkaline radicals (or metallic elements) of the salt of a marketed drug, it has been already marketed overseas, then the application information requirement under Registration Category 3 shall be provided.
17) For the drugs for topical use, in addition to the information required under the relevant Registration Category and Information Items, the information under Information Item 21 should also be submitted; topical absorption test should be conducted, if necessary.
18) When there is an obvious safety concerns in the sustained and controlled released preparations (narrow safety range, significant increase in dosage), animal pharmacokinetic study information compared with the marketed sustained and controlled released preparations or regular preparations should also be provided at single dose.
V Requirement for Clinical Study
1) For the new drugs under Registration Category 1 and 2, clinical trials should be conducted.
ⅰ. Cases of patients for clinical trials should meet the statistical requirement and the minimal cases required.
ⅱ. The minimal cases required (trial group) of clinical trials are as following: 20-30 for Phase I, 100 for Phase II, 300 for Phase III, 2000 for Phase IV,
ⅲ. Phase I clinical trial of the contraceptives should be conducted following this Regulation. In Phase II clinical trial, a randomized controlled clinical study should be conducted on at least 100 pairs of subjects for at least 6 menstruation cycles. In Phase III trial, an open trial on at least 1000 cases for 12 menstruation cycles should be accomplished. In Phase IV trial, variable factors of such kind of drugs should be carefully considered to finish the trial with adequate numbers of cases.
2) For the new drugs under the Registration Categories 3 and 4, human pharmacokinetic study and randomized controlled clinical trials on at least 100 pairs of subjects should be conducted. In the event of more than one indication, cases for each main indication shall not be less than 60 pairs. Human pharmacokinetics study and an open trial on at least 500 cases for 12 menstruation cycles should be accomplished for contraceptives.
Human pharmacokinetics study may be exempted for the following 2 cases:
i) preparation of topcial use with only topical treatment effect.
ii) oral preparation that could not be absorbed.
3) The clinical study for the new drug under Registration Category 5 should be conducted in accordance with the following principles,
i. Bioequivalence trials should be conducted for oral solid preparations on normally 18-24 cases.
ii. When a bioequivalence trial is difficult to be conducted for oral solid preparations or other non- oral solid preparations, clinical trails should be conducted, and cases for the clinical trials should be at least 100 pairs.
iii. For the preparations of sustained and controlled released preparations, controlled human pharmacokinetic study and clinical trials related to therapeutics should be conducted on single dose and repeated doses of the drugs, and the cases for clinical trials should be at least 100 pairs.
iv. Necessary clinical trials should be conducted for injection. When clinical trials are needed, clinical case should be at least 100 pairs for single active component injection, and at least 300 pairs for multiple component injection. For microsphere, microemulsions and liposome, clinical trials should be conducted according to requirement for category 1 and 2.
4) For the oral solid preparations under the Registration Category 6, bioequivalence tests should be conducted on normally 18-24 cases. If the quality of drug needs to be controlled by process and standards, clinical tests should be conducted on normally 100 cases.
5) Application for reduction or exemption of clinical trial should be made during the application of drug registration with detail of reasons and information for reduction or exemption of clinical trials. If a clinical trial is already approved, with exception of the case where reduction or exemption of clinical trial is allowed by this Regulation, other reduction or exemption of clinical trial generally should not be allowed. If there is indeed difficulty to complete the clinical trial, application should be made with detail of the basis and plan for reduction or exemption of clinical trial, where the rational should be justified in term of clinical statistic, status of group of patients in the clinical trials.
6) The comparative drug used for the controlled clinical trails shall be already marketed in China. If the comparative drug must be imported, the approval from SFDA and port institute for drug control is needed. Priority in choosing the comparative drug of positive clinical test should be as following:
i. Drug from the original innovative manufacturer
ii. The same drug of definite clinical test data
iii. Drug of the same active substance and route of administration but different dosage form
iv. Other drug, which is of similar mechanism of action and the same indication.
VI Requirement on Import Chemical Drug
A Requirement of Application Information Items
1) Application Information should be submitted in accordance with the requirement under Table of the Application Information Items of Chemical Drugs. For the application of the new chemical entity not yet marketed in any country, Application Information should be submitted in accordance with the Registration Category 1. For other drugs, Application Information should be submitted in accordance with the Registration Category 3. CTD data as specified by ICH may also be submitted, however, summary general information should be submitted according to requirement of Application Information Items. Drug under Registration Category 1 should be those that are at least in the stage of Phase II Clinical Trials ex-China.
2) Information Item 5, include draft of packaging insert sheet, notes to the draft and the updated literature, the original PI from the manufacturing country, the actual commercial sample of PI used in the manufacturing country and the Chinese translation. The original commercial packaging and labeling should also be provided for Registration Category 6.
3) All the clinical study information used for the market authorization approval in the original manufacturing countries shall be submitted for Information Item 28.
4) All the application information shall be in Chinese with the original text attached, information in other language (ex-English) may be attached as reference. The Chinese translation shall be consistent with the original language.
5) The Chinese translation of quality specification must comply with the format of the National Drug Standards of China.
B Requirement and notes to the Information Item 2, Certified Documents
1) Information Item 2, Certified Documents, includes,
i) Certified Documents, notarized document for the free sale certificate (FSC) issued from the competent authorities of the local country or region where the manufacturer is located, and the GMP Certificate of the manufacturer, and the Chinese translation. Application for the drugs under Registration Category 1, the above Certified Documents can be submitted together with the clinical study report upon the completion of the clinical study in China. However, during the application of Clinical Trails, certified documents of GMP Certificate of the manufacturer issued by local competent drug administration where the drug is manufactured must be provided.
ii) When the registration of a foreign drug manufacturer is conducted by manufacturer’s office in China, copies of Registration Certificate Of Resident Office Of Foreign Enterprise should be provided. When a foreign drug manufacturer authorizes domestic agent to conduct the registration, copies of the authorization document, notarized document and the Chinese translation, as well as the Business License of the domestic agent shall be provided.
iii) Documents and explanations to evidence the patent status and ownership of the drugs of the application, the formula of the drugs, the production technology and process of the drugs, as well as letters of statement stating that the new drug will not infringe upon the patent rights of others.
2) Notes,
i) Certified Documents, notarized document for the FSC and GMP Certificate of the manufacturer should comply with the recommended format by World Health Organization (WHO). The document in other format must be notarized by competent authorities where the manufacturer is located and legalized by the Chinese embassy in the original country.
ii) When the manufacturing site and packaging site separated, the FSC Certified and GMP Certificates of manufacturing and packaging site issued by the corresponding countries should be provided.
iii) In the event that the products not yet approved in the manufacturing country or region, the Certified Documents from the country where the products being marketed and GMP Certificates from this country could be provided. The Certified Documents from the country where the products being marketed and GMP Certificates from this country should be recognized by SFDA.
iv) For the drug substance, the Certified Documents for the approval of the marketing for the drug substance or its preparation issued by the competent authorities of the original manufacturing country, and the GMP Certificate of the manufacturer should be provided. Drug Master File (DMF) or Certificate of Suitability to the Monographs of the European Pharmacopoeia of the drug substance, the Certified Documents for the approval of the marketing for the preparation made of the drug substance issued by the competent authorities of the original manufacturing country, and the GMP Certificate of the manufacturer may also be accepted.
v) To apply for an international multi–center clinical study, report of the manufacture process which ertifies that the drug for clinical study use is manufactured under the GMP must be provided.
vi) For drug substance or preparation administered as food, certified document of GMP Certificate of the drug manufacturer issued by local food and drug administration where the manufacturer is located, ISO9000 quality assurance certificate issued by the competent organization, and / or free sale certificate (FSC) issued from the competent authorities of the local country or region where the manufacturer is located should be provided
C Requirement for the clinical study conducted in China
1) During application of drug never marketed in any country, clinical trials should be conducted according to CT study requirement of Registration Category 1.
2) During application of drug that marketed ex-China but not in China, clinical trials should be conducted according to CT study requirement of Registration Category 3.
3) During application of drug with same route of administration but different dosage form of that of the drug marketed in China, if information item 28 of the application meets the requirement, clinical trials should be conducted according to CT study requirement of Registration Category 5. If information item 28 of the application did not meet the requirement, clinical trials should be conducted according to CT study requirement of Registration Category 3.
4) During application of drug already with National Standards, if information item 28 of the application meet the requirement, clinical trials should be conducted according to CT study requirement of Registration Category 6. If information item 28 of the application did not meet the requirement, clinical trials should be conducted according to CT study requirement of Registration Category 3. No clinical trial is needed for application of drug substance already with National Standards.
5) For the application only for the importation of the drug substance without available National Drug Standards in China, the clinical study shall be conducted with the use of the preparations of the drug substance.
VII Application Information and Requirement for Radioactive Drugs
A Requirement of Information Items
1) For the application of radioactive drugs: the application information shall be prepared in accordance with the corresponding Category of radioactive chemicals, packing case and preparations respectively and the Requirement of Application Information Items. Information Items 22 and 26 may be exempted.
2) For the application of diagnosis radioactive drugs, Information Items 24 and 25 may be exempted.
3) For the application of radioactive chemicals and packing case, Information Item 17 and 18 may be exempted. Information required under preparation should also be provided for the application of packing case.
B Notes to the Information Items
1) Information Item 8 shall be submitted in accordance with the following,
i) Radioactive chemicals: the selection of the production method of the nuclide, irradiation condition, nuclear reaction equation, chemical processing technology after target material eradiated (with the chemical reaction equation and production process attached), detailed operation procedure, possible nuclear impurity introduced, refining (purifying) method, and the specification, standards and analysis of the other chemical reagent (in particular target material), relevant literature and information at domestic and overseas should be provided.
ii) Packing case: the experiment basis for the determining of packing case, the preparation process, route, reaction condition, operation steps and quality standards of all components of the packing case shall be provided. If any components was made by the manufacturer, then the basis of determining of the detailed synthesis route, synthesis process flow, reaction equation, chemical equation, reaction condition, operation procedure, material input, output ratio, and possible impurity introduced and mixed, quality control of the intermediate products at each steps, refining (purifying) method of finished product, quality standard of the raw material, literature and information at domestic and overseas shall be provided.
iii) Preparation: basis to choose the formula of the preparation, preparation process, reaction condition, operation procedure, refining or purifying method, quality standards and analysis and test data of raw material, and literature and information at domestic and overseas shall be provided.
2) Information Item 9 shall be submitted in accordance with the following,